


















 89 / 210
89 / 210

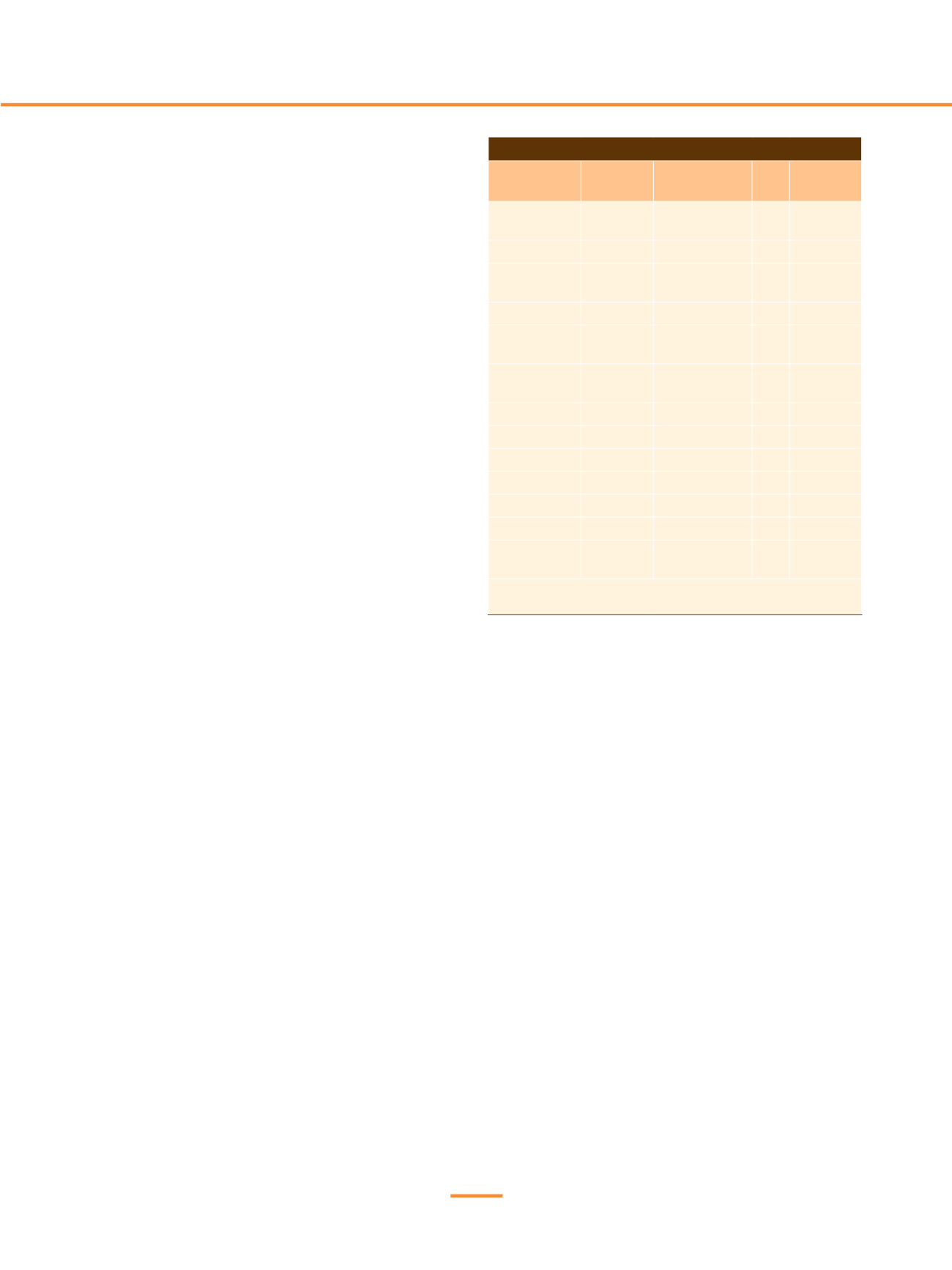
Sesión Plenaria
89
Diátesis hemorrágica
SP-005
Identificación de 211 sujetos con
deficiencia de FXI portadores de
10 mutaciones y seguimiento durante
20 años: implicaciones epidemiológicas,
diagnósticas y clínicas
Esteban J. (1), De la Morena-Barrio M. E. (2,3), Salloum-Asfar
S. (1), Padilla J. (1), Miñano A. (1), Campillo I. (1), Martínez-
García M. Á. (1), Beltrán V. (1), Soria J. M. (4), Ferrer F. (2,3),
Vidal F. (5,6), Martín L. (4), Vicente V. (2,3), Corral J. (2,3)
(1) Hospital Virgen del Castillo. Yecla, Murcia. (2) Servicio de Hematología y
Oncología Médica. Hospital Universitario Morales Meseguer. Centro Regional
de Hemodonación. Universidad de Murcia. IMIB-Arrixaca, Murcia. (3) Grupo
de investigación CB15/00055. Centro de Investigación Biomédica en Red de
Enfermedades Raras (CIBERER). Instituto de Salud Carlos III (ISCIII). Madrid.
(4) Unitat de Genòmica de Malalties Complexes. IIB-Sant Pau. Barcelona.
(5) Coagulopaties congènites. Banc de Sang i Teixits. Barcelona. (6) Unitat de
Diagnòstic i Teràpia Molecular. Vall d’Hebron Institut de Recerca. Universitat
Autònoma de Barcelona (VHIR-UAB). Barcelona
Introducción:
Recientes evidencias sustentan la relevancia
hemostática del FXI. Su deficiencia, considerada un desorden
raro, tiene escasa importancia hemorrágica pero podría proteger
contra eventos trombóticos.
Objetivo:
Caracterizar la deficiencia de FXI y sus conse-
cuencias clínicas en un área de 60.000 habitantes de la Región
de Murcia.
Métodos:
En 20 años (1994-2014) se realizaron 324.764
pruebas de TTPa en 51.366 pacientes; se seleccionaron 1.700 que
presentaron un TTPa: r prolongado (> 1,3). Descartados los no
confirmados, anticoagulados y los anticoagulante lúpico, se estu-
diaron los factores de la vía intrínseca.
El análisis genético incluyó secuenciación (Sanger y NGS),
MLPA y genotipado con sondas Taqman. El FXI plasmático se
estudió mediante Western-blot.
Resultados:
Identificamos 211 casos con deficiencia de FXI,
7 en homocigosis, 3 en heterocigosis compuesta y 201 en hetero-
cigosis pertenecientes a 42 familias no relacionadas. Destacamos
3 mutaciones fundadoras: p.Cys56Arg (descrita en población
vasco-francesa), p.Cys416Tyr y p.Glu565Lys identificadas en 23,
7 y 6 casos índice, respectivamente. Además, otras 8 mutaciones
diferentes, 3 de ellas no descritas previamente, se detectaron en
7 casos índice. Dos mutaciones, una de ellas nueva, provocaron
deficiencia CRM+ (proteína variante en plasma pero sin actividad
funcional)
(Tabla 1)
.
El TTPa no estaba prolongado (ratio < 1,3) en 51/211 casos
con deficiencia de FXI (24%).
La incidencia de sangrado espontáneo fue escasa (13,6%)
y muy moderado (epistaxis). Únicamente el 5,8% de las 207
intervenciones quirúrgicas y el 4,2% de los 118 partos presenta-
ron complicaciones hemorrágicas, y solo 5 pacientes precisaron
transfusiones. La mayor incidencia de hemorragias se produ-
jo en intervenciones que afectaban a tejidos con alta actividad
fibrinolítica. La tasa de eventos hemorrágicos fue similar, in-
dependientemente de la administración profiláctica de plasma
fresco congelado (PFC) (realizada en 58 ocasiones). Sin embar-
go, el PFC causó TRALI en dos pacientes. Nueve pacientes, con
deficiencia de FXI bajo tratamiento anticoagulante (mayorita-
riamente por fibrilación auricular), no sufrieron complicaciones
hemorrágicas.
Seis casos presentaron infarto agudo de miocardio y 8 ictus
isquémico. Sin embargo, solo 2 casos sufrieron trombosis venosa
(a pesar de que 16 pacientes también eran portadores del FV Lei-
den o protrombina G20210A).
Conclusiones:
Identificamos una alta incidencia de defi-
ciencia de FXI con notable variabilidad genética en un área
de la Región de Murcia. Estos resultados, junto con la eleva-
da incidencia de falsos negativos del TTPa y la escasa clínica
hemorrágica de portadores, sugieren que la incidencia de este
desorden en población general está subestimada y podría al-
canzar el 1%.
La deficiencia de FXI no incrementa notablemente el riesgo
hemorrágico, ni siquiera si se combina con tratamiento anticoa-
gulante oral. La administración de PFC no supone ningún benefi-
cio, pero puede tener importantes efectos adversos.
La deficiencia de FXI, particularmente en heterocigosis, no
parece proteger de la trombosis arterial, pero podría proteger del
desarrollo de trombosis venosa. Estos resultados, compatibles
con los modelos animales y ensayos clínicos de silenciamiento de
FXI, y junto a la alta incidencia de la deficiencia de FXI, apoyan
Tabla 1.
Alteraciones del F11 identificadas en nuestro estudio
Alteración F11
Mutación
(HGMD)
Estatus
genético
N
Tipo de
deficiencia
p.Cys56Arg
CM020681
Homocigosis
Heterocigosis
3
105
CRM-
p.Cys416Tyr
CM053240 Heterocigosis 19
CRM-
p.Cys56Arg &
p.Cys416Tyr
CM020681
CM053240
Het compuesta 1
CRM-
CRM-
p.Glu565Lys
CM051917 Heterocigosis 27
CRM-
p.Pro538Leu
CM051916
Homocigosis
Heterocigosis
4
30
CRM+
p.Lys536Asn &
p.Cys599Tyr
CM002953
Nueva
Het compuesta 2
CRM-
CRM+
p.Lys536Asn
CM002953 Heterocigosis
1
CRM-
p.Cys599Tyr
Nueva
Heterocigosis
1
CRM+
p.Thr322Ile
CM950373 Heterocigosis
3
CRM-
p.Arg268Cys
CM035499 Heterocigosis
2
CRM-
c.325G>A
CS081910 Heterocigosis
4
CRM-
p.Ile426Thr
Nueva
Heterocigosis
4
CRM-
Duplicación
exones 8 y 9
Nueva
Heterocigosis
5
CRM-
CRM: del inglés cross reactive
material.
CRM+: con proteína variante en circulación.
CRM-: sin proteína variante en circulación. HGMD:
Human mutation database.