


















 28 / 210
28 / 210

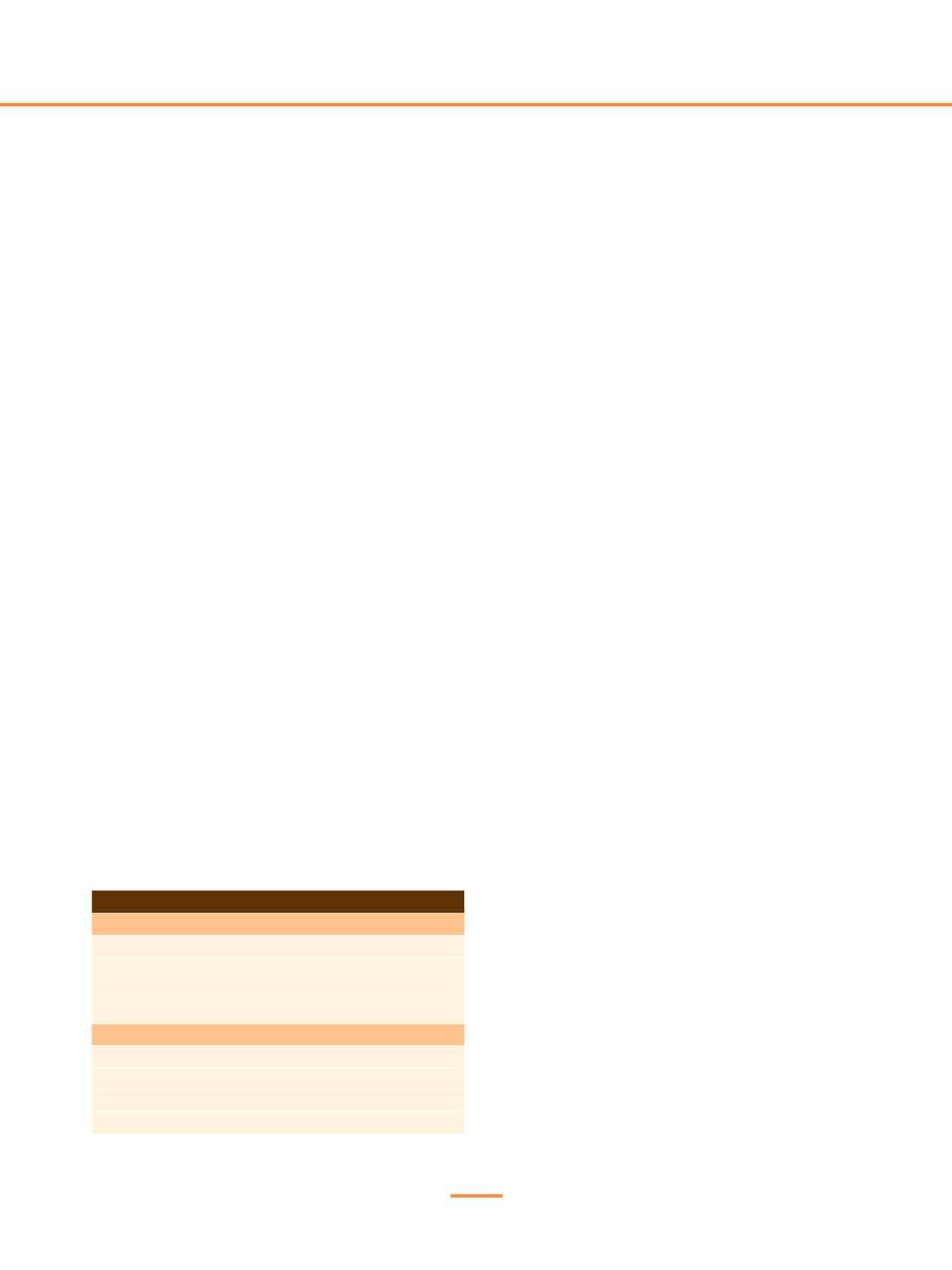
XXXII Congreso Nacional de la Sociedad Española de Trombosis y Hemostasia
28
Desde las primeras descripciones de la hemostasia realizadas
por Morawitz, a principios del siglo XX, han sido muchos los
conocimientos adquiridos sobre este complejo sistema donde la
hemostasia y la trombosis van de la mano y su visión conjunta
es imprescindible para mejorar el control de las manifestaciones
clínicas en estos pacientes. Las novedades en las coagulopatías
congénitas y las que se desarrollan para un futuro cercano están
focalizadas a mejorar el diagnóstico, prevenir las complicaciones
y avanzar en la posible curación de la enfermedad. En esta últi-
ma década se han aplicado todos los avances tecnológicos usados
para la investigación en hemostasia: la estructura química de las
proteínas, el estudio del ADN, la terapia génica y, por último, la
medicina regenerativa
1
.En la
Tabla 1
se presentan los principales
hitos alcanzados y los retos que se desea conseguir.
La disponibilidad de sistemas automatizados para el estu-
dio de la hemostasia permite detectar mínimas anomalías me-
diante distintas técnicas; no obstante, es imprescindible obtener
una historia clínica detallada, personal y familiar, para orientar
correctamente una enfermedad hemostática congénita. Las prue-
bas globales de coagulación, en especial la del test de genera-
ción de trombina, podrán ser de gran utilidad si se consigue su
estandarización para pacientes con coagulopatías, en especial en
pacientes con hemofilia e inhibidor, dado que precisan de trata-
miento con agentes baipás o cuando coexiste un riesgo trombóti-
co asociado a la deficiencia hemostática
2
.
La enfermedad de von Willebrand es la coagulopatía congé-
nita más prevalente y, a pesar de ello, continúa siendo la que más
dificultades genera para su identificación. Actualmente, la dispo-
nibilidad de índices clínicos propios, de pruebas de laboratorio
específicas y la identificación de las anomalías genéticas subya-
centes abren un nuevo campo de estudio de esta enfermedad cuya
expresión clínica es muy dispar. El registro español sobre el perfil
clínico y molecular correlaciona los datos fenotípicos y genotípi-
cos a partir de una metodología de estudio centralizada. En él se
ha incluido un total de 556 pacientes de 38 hospitales, lo que ha
permitido clasificar correctamente el tipo en 480 pacientes y per-
filar adecuadamente el fenotipo y genotipo en el 96,5% de ellos.
Cabe destacar que las nuevas técnicas de secuenciación han per-
mitido llevar a cabo el estudio molecular con un coste accesible
3.
Las otras coagulopatías congénitas (deficiencias de fibrinóge-
no o de factores II, V, V y VIII asociados, VII, X, XI y XIII y la
de vitamina K) representan un 3-5%. Se estima una incidencia de
1 por 500.000 para la deficiencia de factor VII y de 1-2 casos por
3 millones para la de factor XIII. Los resultados de los estudios
epidemiológicos de laWorld Federation of Hemophilia y de la red
europea (EN-RBD) han permitido identificar las características
de estos pacientes, clasificarlos según el fenotipo hemorrágico,
su distribución, las manifestaciones clínicas, los niveles mínimos
hemostáticos y el estudio molecular; las mutaciones de aminoáci-
dos
(missense)
están presentes en el 50-80% de las identificadas.
En la actualidad todas las deficiencias disponen de tratamiento
sustitutivo específico de origen plasmático, con excepción de la
de factor V, la cual solo dispone de plasma y la de factor XIII de
proteína recombimate para el subtipo FXIII-A. Existen recomen-
daciones generales sobre los niveles mínimos hemostáticos, pero,
debido a su baja prevalencia, es necesario continuar los registros
y estudios colaborativos
4
.
Por sus manifestaciones clínicas y por ser la más prevalente
de las coagulopatías graves, el tratamiento de la hemofilia ha vi-
vido un giro copernicano (del sustituto plasmático a las proteínas
recombinantes), lo cual ha permitido mejorar la calidad de vida
de los enfermos hemofílicos. No obstante, debido a su elevado
coste, la disponibilidad de este tratamiento solo está al alcance de
un número limitado de ellos.
El tratamiento profiláctico, de largo recorrido histórico en
algunos países como Suecia y Holanda, ha demostrado su supe-
rioridad frente al tratamiento según demanda, en estudios alea-
torizados. A tenor de la gran variabilidad de la vida media de los
factores VIII y IX, los estudios famarcocinéticos son básicos para
disponer en cada paciente de un modelo individualizado sobre
la cinética del factor deficitario
5
. Estudios recientes remarcan
la influencia del sistema sanguíneo ABO y los niveles de factor
von Willebrand sobre la farmacocinética del factor VIII. La do-
sis profiláctica debe adaptarse a los parámetros farmacocinéticos,
teniendo en cuenta además el fenotipo hemorrágico y el estilo de
vida del paciente. La profilaxis previene el desarrollo de la artro-
patía y son necesarios estudios de imagen que permitan detectar
su inicio y evolución. La resonancia magnética ha sido de gran
Avances en coagulopatías congénitas
C. Altisent
Unitat d’Hemofília. Hospital Universitari Vall d’Hebron. Barcelona
Tabla 1.
Hitos y retos
Hitos alcanzados
Diagnóstico molecular
Profilaxis de la hemofília en edades tempranas
Aumento de la esperanza de vida
Nuevas moléculas recombinantes de semivida plasmática larga
Retos a conseguir
Disponibilidad universal de los fármacos sustitutivos
Prevención y erradicación del inhibidor
Control de la comorbilidad
Curación de la enfermedad